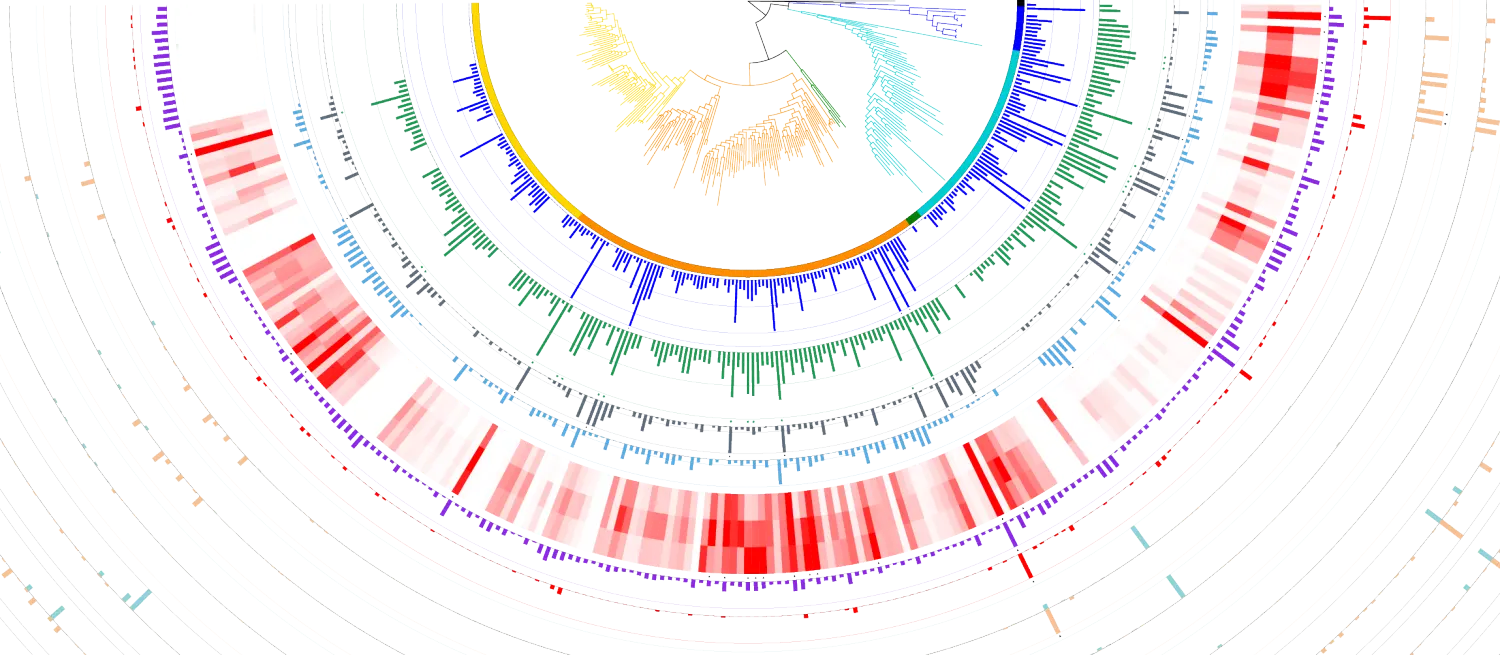
分子层面对生物的研究,在个体水平上主要是看单个基因的变化以及全转录本的变化(RNA-seq);在对个体的研究的基础上,开始了群体水平的研究。如果说常规的遗传学主要的研究对象是...
 发简信
发简信
IP属地:澳门


分子层面对生物的研究,在个体水平上主要是看单个基因的变化以及全转录本的变化(RNA-seq);在对个体的研究的基础上,开始了群体水平的研究。如果说常规的遗传学主要的研究对象是...
您好,我跟您一样也出现了【因为找不到数据库都有哪些物种的,这些信息在organism.db数据库中】这个报错信息,请问这个要怎么解决呢?求指教,感谢🙏
KOBAS3-docker终于跑起来了。前言:Docker安装成功了Kobas下载成功了,也load成功了无意间的操作,竟然跑成功了。呵呵 在成功跑起来之前,一直没有运行映射命令。 如果没有运行上面的映射命令。运行...
好的,感谢!@儒雅随和王小板
比对软件STAR的使用在之前的学习和练习里,比对这一步我使用过bowtie2(DNA比对)和hisat2(RNA-seq比对),现在学习另一个很火的软件:STAR。STAR能够发现非典型拼接和嵌合...

大致的流程如下:BBtools --》 Trinity(v2.8.5) --》 Transdecoder --》Trinotate (3.2.1) 1. 合并左右read...
请问,我用trinity自带脚本abundance_estimates_to_matrix.pl构建转录本-基因表达矩阵时,报错Use of uninitialized value为什么呢?😭
对trinity组装得到的转录本进行表达矩阵构建前提已经获得了trinity生成的.fasta文件(trinity_out_dir_all.Trinity.fasta) 在此,我们采用的是Trinity自带的脚本进行定量计...
1分析步骤 1.1序列延伸(inchworm)--虫 -将reads切割为 k-mers(k bp长度的短片段)-利用Overlap关系对k-mers进行延伸(贪婪算法)-输...
前面三节,我们得到了Trinity拼接的Fasta 文件(Trinity.fasta)以及通过Bowtie2将Fastq中的Reads进行回贴到Trinity.fasta文件...
请问一个很简单的问题,怎么把样品写到一个Sample_text中呢😭
转录组分析实战第三节: RESM对Trinity得到的转录本进行定量前面三节,我们得到了Trinity拼接的Fasta 文件(Trinity.fasta)以及通过Bowtie2将Fastq中的Reads进行回贴到Trinity.fasta文件...
大神,请教一下,后续进行转录组定量的时候,是用clean data还是用bowtie2回帖后比对生成的文件呢?
转录组分析(5) - 无参转录组拼接(illumina)目的 NGS测序得到的短序列(read)存储于Fastq文件,在经过DNA建库和测序之后,文件中不同read之间的顺序就全部丢失了。因此,Fastq文件中紧挨着的两条read...
您好,请问我在筛选同一基因的最长转录本作为unigene时,出现note:longest transcript isn't always the best transcript!... consider filtering based on relative expression support 。是不是不用管它呀?
无参转录组序列组装 — Trinity转录组分析策略 转录组分析策略根据有无参考转录组可以分为两类: 有参比对—定量—差异分析—功能富集分析等 无参组装—定量—差异分析—功能富集分析等image 无参考转录组序列...
您好,我想请教下,去冗余方法(1)筛选同一基因的最长转录本作为unigene;(2)软件:TGICL、CAP3通过聚类筛选unigene。是不是两者选一个使用就好啊?
无参转录组序列组装 — Trinity转录组分析策略 转录组分析策略根据有无参考转录组可以分为两类: 有参比对—定量—差异分析—功能富集分析等 无参组装—定量—差异分析—功能富集分析等image 无参考转录组序列...
请教下,做无参转录组分析,一共6*3=18个样品,是把所有样品放在一起组装成一个转录本吗?
mRNA-seq学习(六):转录本的de novo组装1. 组装 2. 结果 按照1.中的参数设置(内存和CPU)和数据量(4.9G的fasta),耐心等待6小时就能完成组装了。在这之后就是比对过程了,再然后的差异分析和富集分析...