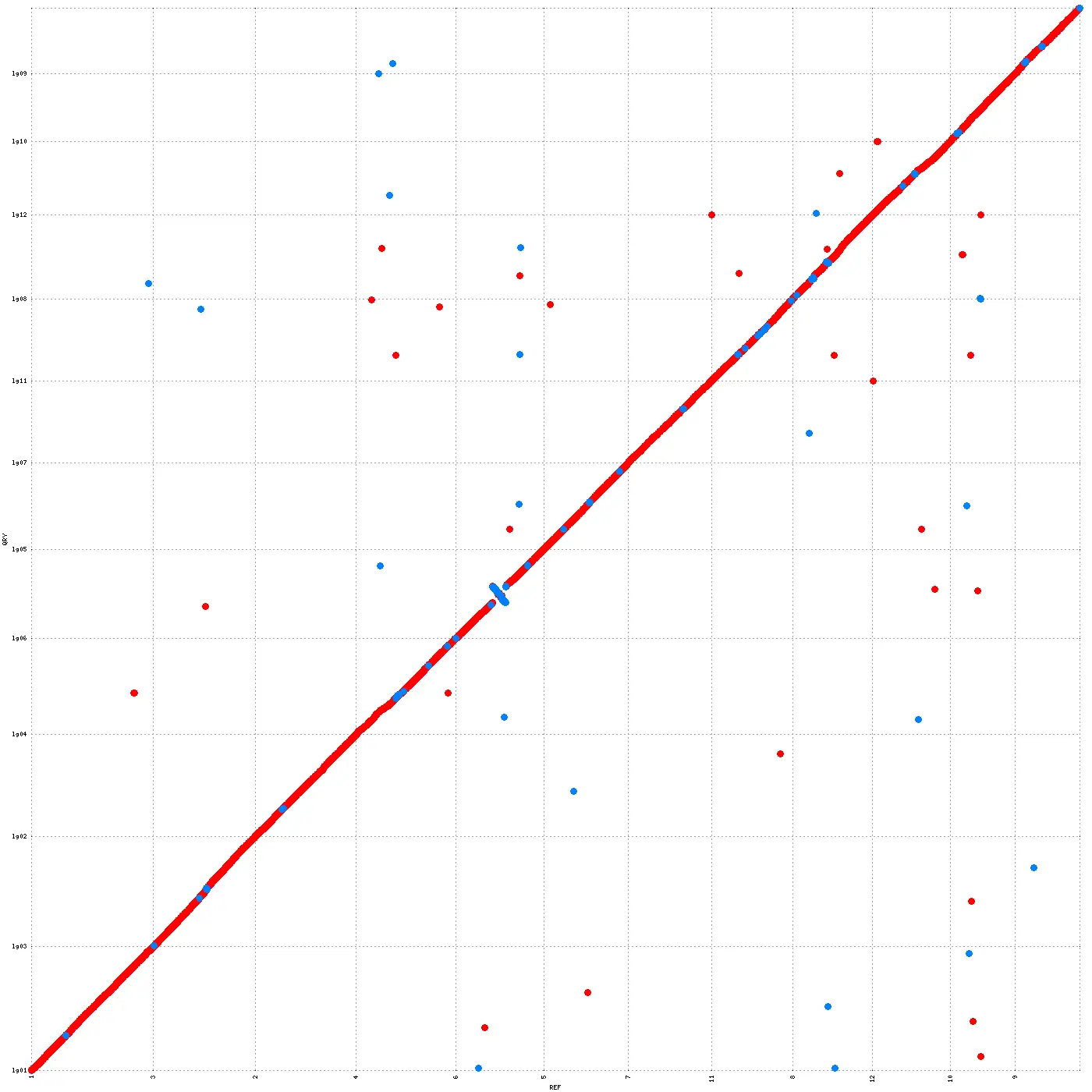
WGCNA图文详解(七)-eigengene network的可视化 还是老习惯,给出官网教程,至于你是看还是不看,它就在那里,等着你的深入研究~ https://horva...
 发简信
发简信
IP属地:北京


WGCNA图文详解(七)-eigengene network的可视化 还是老习惯,给出官网教程,至于你是看还是不看,它就在那里,等着你的深入研究~ https://horva...

在上一篇帖子上完成了表达量数据和表型数据的整理,这篇文章将进行网络构建和模块检测。 WGCNA(1):R包安装及数据导入清洗 - 简书 (jianshu.com)[https...

WGCNA:加权基因共表达网络分析,简而言之,就是将基因划分为若干个模块,探究与表型数据与基因模块之间的相关关系,并找到模块中的核心基因。 适用于复杂的数据模式,推荐5组(或...
在前两篇帖子中介绍了数据的导入清洗,以及一步法构建网络,这篇文章将介绍网络构建的第二种方法,分步法。WGCNA(1):R包安装及数据导入清洗 - 简书 (jianshu.co...
作者你好呀,想请问一下为什么要去掉Q矩阵的最后一列呢?
重测序分析(16)GWAS分析实操(2)gwas_emmax_Q使用Q矩阵作为协变量使用emmax进行GWAS分析 数据准备 1.上个推文中vcf转的tped文件snp_filter.mapsnp_filter.nosexsnp_filt...


数据处理 VCF转为 rrBLUP {-1,0,1} 格式 rrBLUP可识别的基因型格式为 {-1,0,1} (行头为marker,列为sample),因此需要对基本数据...
想请教一下怎么进行k折交叉验证呢
【GS模型】全基因组选择之rrBLUP[toc] 1. 理论 rrBLUP是基因组选择最常用的模型之一,也是间接法模型的代表。回顾一下,所谓间接法是指:在参考群中估计标记效应,再结合预测群的基因型信息将标记效应累...

GATK的HaplotypeCaller 应该是目前最常用的变异检测软件,尤其是在人类基因组上。不过HaplotypeCaller的速度相对于其他软件,例如bcftools,...

以GATK流程为例,简单来说,SNP Calling主要包括以下几步: 1. 给参考基因组建立索引:samtools faidx、bwa index, gatk Create...
GATK4.0 和之前的版本相比还是有较大的不同,更加趋于流程化。 软件安装 点击此处 查看最新版本 GATK 简单说明 GATK分析简要流程 所需数据 ref.fa rea...
使用BWA+samtools+gatk4.0进行SNP calling 首先下载数据eg: 01.align 首先定义相关用到的变量,然后使用baw创建索引之后进行map。 ...

一.软件下载安装 🔸bwa:https://github.com/lh3/bwa[https://github.com/lh3/bwa]安装及命令详解参考:http://st...
一、简介 md5sum计算检验MD5效验码。MD5算法常常被用来验证网络文件传输的完整性,防止文件被人篡改。MD5全称是报文摘要算法(Message-Digest Algor...
annovar简介 在我们做重测序,或者比较基因组学分析的时候,我们往往在call snp或者indel的时候,需要对我们的变异位点进行注释,而annovar官网提供的注释库...